
This is a tutorial to use the iMODFIT molecular fitting tool in Chimera. As a user, you will be able to load the plugin and perform a flexible fitting between PDB's and your density map. Just follow the instructions and enjoy : -)
Loading the plugin
To load the plugin in Chimera:
- If you don't already have a directory on your computer that is specific for external plugins for chimera, create one called 'Plugins' wherever you want.
-
Download the plugin from here.
-
Uncompress the file in the 'Plugins' directory.
- In Chimera, go to Favorites → Preferences, from Category select Tools and add the 'Plugins' directory containing the ADP EM folder to the Locations.
The iMODFIT module will be under the Tools → EM Fitting menu.
Using the plugin
When loaded, you will see this new window:
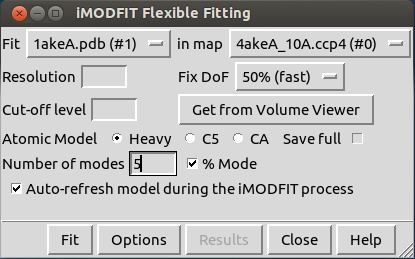
In the first row, you must choose the atomic structure and the EM map to be fitted from those already loaded in Chimera.
There are three required parameters to start the flexible fitting process with iMODFIT plugin:
- Resolution: Nominal resolution of the selected map (in Å).
- Fix DoF: Percentaje of randomly fixed dihedral coordinates. Example: set 50% to randomly fix the half of the dihedral coordinates, i.e. the Phi and Psi canonical dihedral angles. (Rotational/Translational coordinates are always mobile). Values can be None, 50% (fast), 70% (faster) and 90% (fastest).
- Cut-off: Is the density threshold value for the experimetal map. All density levels below this value will be not considered. You can get this value from the level parameter showed in the Volume Viewer to adjust with more precision your map density.
- Atomic Model: coarse-Grained model:
- - Heavy → employ all heavy-atoms present in the PDB, i.e. all non-hydrogen atoms.
- - C5 → five pseudo-atoms; it uses NH, Cα, CO, Cβ and virtual mass located at the mass center.
- - CA → use a single Cα atom per amino acid (N and C atoms are always required to define dihedral angles).
- Save full: Enable this check-box to generate a complete model from the reduced representations (C5 and Cα). This may be very useful in case not all the heavy atoms are present in your input PDB. For example, using this option, iMODFIT will internally employ a coarse-grained Cα representation but, at the same time, produce models with all heavy atoms present in the PDB.
- Number of Modes: Number of modes used, either number [1,N] , or percentage (if check-box is selected). Percentages between 2 and 10% are generally fine, but if you don't know what is this, please, use the 5%.
In addition to these parameters, some extra features had been provided for expert users that know how to use the iMODFIT command-line tool. These parameters can be introduced in the Options button, which displays a new panel to insert them:
- Introduce options: Field to insert command line options. They are all explained in the iMODFIT documentation (see Further Information). Example: --delta_save 0.2
- Rediagonalization: RMSD increment to trigger re-diagonalization (in Å). (default=0.1)
Once you have inserted all the parameters you want for the fitting, simply click Fit and enjoy. You will see the process log in a new window and, when it is finished, you could check the results in the correspondet panel.
Further Information
You can expand your knowledge on the use of the iMODFIT tool in the following links:
Main: http://chaconlab.org/hybrid4em/imodfit
Getting Started: http://chaconlab.org/hybrid4em/imodfit/imodfit-intro
Tutorial: http://chaconlab.org/hybrid4em/imodfit/imodfit-tutorial
Tips&Tricks: http://chaconlab.org/hybrid4em/imodfit/imodfit-tips
Other Resources: http://chaconlab.org/hybrid4em/imodfit/imodfit-other
Download: http://chaconlab.org/hybrid4em/imodfit/imodfit-donwload
Credits
The plugin was developed by Pablo Solar for Pablo Chacón's group (chaconlab). Acknowledgements go to Pablo Chacón and José Ramón López Blanco, CSIC, for sharing their time with me, wisdom and friendliness, and to Tom Goddard, UCSF, for providing useful insights.
Citation
If you find this plugin useful for your research project, please cite:
- iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates (2013). López-Blanco J.R. and Chacón P. JSB 184(2):261–270

