
The ADP_EM Tutorials are practical guides for researchers who want to use ADP_EM to perform multiresolution docking. They include working examples and simple instructions. Before you go through the tutorial, first get the corresponding files from download zone. The following tutorials are currently available:
As first exercise let’s dock the GroEl subunit into the whole GroEL-GroES map. The GroEL-ATP from E.coli was taken from The Macromolecule Structure Database (EMD). The map with EMD ID 1047 emd1047.ccp4 was downloaded from EMD (It was also pre-processed to remove discontinuous density using voledit tool of SITUS package, floodfill at 0.060). The docking exercise will consist in the localization of a GroEL subunit (chainA.pdb extracted from 2C7E pdb) and their related symmetric copies. First download, uncompress and untar the corresponding single tutorial file. This file contains all the necessary files to perform the tutorial. I recommend create a new working directory, so all the output files will be stored on it. To perform the docking just prompt:
where 16 corresponds to the harmonic bandwidth used, 14.9 is the resolution in angstroms, 0.06 the density cutoff used and 18 the number of fits saved. Here is the screen output:
adp_em>
adp_em> ADP_EM
adp_em>
adp_em> Single PDB
adp_em> Laplacian Kernel Filtering
adp_em> Search Mode: Mask
adp_em> Save 20 best fits
adp_em> Sampling-> Rot: 11.250 (bw=16) Trans: 3.725
adp_em>
adp_em>
adp_em> Step 1: Processing EM-map & pdb...
adp_em>
adp_em> 3DMAP ../emd1047.ccp4
adp_em> Resolution 14.900000 cutoff 0.060000
adp_em> Initial dimensions 128x128x128
adp_em> Grid Size 1.862x1.862x1.862
adp_em> Mass center 40.999x41.001x44.586
adp_em> Radii E 8 54 I 6 40 Z 0 0
adp_em> Radii E 11 55 I 0 0 Z 12 56
adp_em> Final dimensions 85x85x89 voxels
adp_em>
adp_em> PDB ../chainH.pdb
adp_em> DIM: Low pass 50x49x58
adp_em> Radii E 9 31 I 0 0 Z 10 32
adp_em> Final dimensions 52x51x60
adp_em>
adp_em> Step 2: Search...
adp_em>
adp_em> Trans limits min 0.000 max: 46.000 init 0.000
adp_em> Explored 16737 (mask 134001 not 66468): time 244.02 sec
adp_em> Max peaks 100 (allowed 100)
adp_em> Peaks found 18 (from 100 peaks) time: 251.29 sec
adp_em>
adp_em> Total searching time 252.30 sec
adp_em>
adp_em>
adp_em> Step 3: Saving Solutions...
adp_em>
adp_em> -------------------------------------------------------------------------
adp_em> Psi Theta Phi X Y Z Corr
adp_em> -------------------------------------------------------------------------
adp_em> 1 315.80 -0.00 304.65 52.00 18.00 64.00 0.248
adp_em> 2 341.06 -0.00 228.44 30.00 20.00 64.00 0.248
adp_em> 3 344.72 -0.00 63.03 44.00 68.00 64.00 0.239
adp_em> 4 352.64 -0.00 116.45 22.00 60.00 64.00 0.238
adp_em> 5 313.56 -0.00 201.01 16.00 38.00 64.00 0.236
adp_em> 6 241.29 -0.00 72.41 66.00 36.00 64.00 0.235
adp_em> 7 353.09 -0.00 15.62 64.00 58.00 64.00 0.235
adp_em> 8 316.95 180.00 301.91 58.00 22.00 24.00 0.223
adp_em> 9 315.23 180.00 44.85 58.00 62.00 24.00 0.221
adp_em> 10 27.06 180.00 164.44 35.86 66.75 24.04 0.221
adp_em> 11 243.06 180.00 285.23 68.00 42.00 24.00 0.221
adp_em> 12 358.67 180.00 192.71 18.55 53.01 23.97 0.220
adp_em> 13 290.07 172.32 227.73 36.00 18.00 24.00 0.218
adp_em> 14 296.26 172.33 176.39 18.00 32.00 24.00 0.216
adp_em> 15 82.25 6.49 284.40 60.00 58.00 64.00 0.182
adp_em> 16 81.49 5.67 25.02 22.00 56.00 64.00 0.180
adp_em> 17 83.58 5.67 330.74 40.00 66.00 64.00 0.178
adp_em> 18 45.56 173.93 336.97 36.00 14.00 24.00 0.170
adp_em> --------------------------------------------------------------------------
adp_em>
adp_em> Saved in adpEM[1-18].pdb files
adp_em> Total Time 255.63 sec
adp_em>
adp_em>
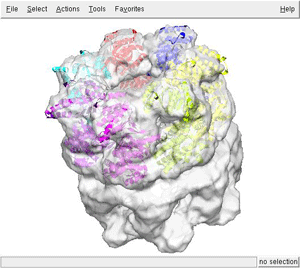
For visualizing the results use your favorite program. Here we display superimposed the 7th first best fittings (files from adpEM0001.pdb to adpEM0007.pdb) with the GroEL-GroES map using chimera
 |
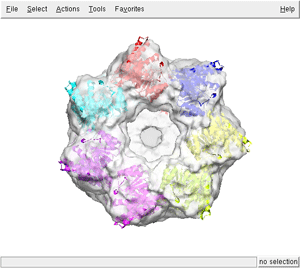 |
As you can see, the heptameric ring has been properly reconstructed.
If you are interesting in docking multiple atomic structures into single EM density map this is your tutorial. As an example we are going to dock a native structure (used to generate the target density maps), 300 homology models, an a template structure (remote homolog used to generate the models) into a simulate EM map. The homology models and the maps has been downloaded from http://salilab.org/modem/modem_benchmark.tar.gz. Since ADP_em does not support MRC format, the maps have been converted to Situs format (ccp4 can be used as well). The tutorial file includes all the models and the EM maps only for 1DXT case (the original Sali's benchmark includes another 7 test cases than can be processed in a equivalent way). Please follow the step by step instruction showing how to perform this multiple docking case.
1) Create column PDB list. As a required input the user must provide a one column ASCII file with the file names of the pdbs to be docked. In our case all the models are stored in a single directory named 1dxt. To generate such list you can take advantage of the "find" Unix tool. As in previous tutorial, create a new directory and work on it.
With previous command we have all the pdb names stored on the file "listpdb" in a single column format .
2) To run the program:
where -m is mandatory flag for multiple docking (otherwise listpdb will be interpreted as a single input file, and the program will crash), 16 is the bandwidth, 10 the resolution of the map, and 0.0 the density cut-off. Since the atomic models accounts for all the density of the map the fitting criteria is set to simple density cross-correlation (-f 0 option). Here is the corresponding output:
adp_em>
adp_em> ADP_EM
adp_em>
adp_em> Multiple PDBs (302 files)
adp_em> No Filtering
adp_em> Search Mode: Mask
adp_em> Save 50 best fits
adp_em> Sampling-> Rot: 11.250 (bw=16) Trans: 2.000
adp_em>
adp_em>
adp_em> Step 1: Processing EM-map & pdb...
adp_em>
adp_em> 3DMAP ../1dxt-10A.sit
adp_em> Resolution 10.000000 cutoff 0.000000
adp_em> Initial dimensions 64x64x64
adp_em> Grid Size 1.000x1.000x1.000
adp_em> Mass center 31.000x32.000x33.000
adp_em> Radii E 21 38 I 0 0 Z 22 39
adp_em> Radii E 21 38 I 0 0 Z 22 39
adp_em> Final dimensions 62x65x65 voxels
adp_em>
adp_em> Step 2: Exaustive search...
adp_em>
adp_em>
adp_em> PDB ../1dxt/1dxt.pdb 1/302
adp_em> DIM: Low pass 56x61x60
adp_em> Radii E 17 35 I 0 0 Z 18 36
adp_em> Final dimensions 56x61x60
adp_em> Trans limits min 0.000 max: 21.000 init 0.000
adp_em> Explored 439 (mask 3471 not 33320): time 4.34 sec
adp_em> Max peaks 100 (allowed 100)
adp_em> Peaks found 12 (stored 12 (max 100) time: 10.35 sec
adp_em>
adp_em> PDB ../1dxt/1hbg.pdb 2/302
adp_em> DIM: Low pass 59x61x50
adp_em> Radii E 16 31 I 0 0 Z 17 32
adp_em> Final dimensions 59x61x50
adp_em> Trans limits min 0.000 max: 22.000 init 0.000
adp_em> Explored 639 (mask 5117 not 33120): time 5.79 sec
adp_em> Max peaks 100 (allowed 100)
adp_em> Peaks found 15 (stored 27 (max 100) time: 11.18 sec
...
adp_em> Total searching time 3920.000000 sec
adp_em>
adp_em>
adp_em> Step 3: Saving Solutions...
adp_em>
adp_em> -------------------------------------------------------------------------
adp_em> Psi Theta Phi X Y Z Corr
adp_em> -------------------------------------------------------------------------
adp_em> 1 259.12 -0.00 101.62 30 32 34 0.986 ../1dxt/1dxt.pdb
adp_em> 2 150.41 141.53 121.65 32 32 32 0.962 ../1dxt/model34.pdb
adp_em> 3 144.19 132.59 114.67 32 32 32 0.960 ../1dxt/model54.pdb
adp_em> 4 142.22 154.84 127.22 30 32 34 0.958 ../1dxt/model195.pdb
adp_em> 5 171.19 146.19 142.90 32 32 32 0.955 ../1dxt/model94.pdb
adp_em> 6 174.37 131.21 129.78 32 32 32 0.953 ../1dxt/model51.pdb
adp_em> 7 157.61 141.37 126.43 32 32 32 0.952 ../1dxt/model60.pdb
adp_em> 8 146.24 136.78 112.96 32 32 32 0.952 ../1dxt/model35.pdb
adp_em> 9 167.50 138.04 134.76 32 32 32 0.952 ../1dxt/model97.pdb
adp_em> 10 151.20 144.26 118.80 32 32 32 0.951 ../1dxt/model18.pdb .....
adp_em> 49 173.69 148.52 142.39 32 32 32 0.941 ../1dxt/model71.pdb
adp_em> 50 173.13 127.89 135.25 30 32 34 0.941 ../1dxt/model81.pdb
adp_em> --------------------------------------------------------------------------
adp_em>
adp_em> Saved in adpEM[1-50].pdb files
adp_em> Total Time 3921.000000 sec
adp_em>
adp_em>
4) Inspect the results adpEM[1-50].pdb with your favorite viewer. You can for example compare the obtained docking poses of the native (1dxt.pdb) and the best fitting model (model34.pdb), which correspond to the first (adpEM0001.pdb) and second best fit (adpEM0002.pdb), respectively.
 |
Note the high similarity of the best fitting model with the original structure used for generate the map.
3) At adp_em_multi.log file you can find the 10 best fits of each model. If you want to build a table with the best fit of each homology model, just run the following commands:
sort -r -k 11 temp > results10
>more results10
1 259.12 -0.00 101.62 30.00 32.00 34.00 0.986 ../1dxt/1dxt.pdb
1 150.41 141.53 121.65 32.00 32.00 32.00 0.962 ../1dxt/model34.pdb
1 144.19 132.59 114.67 32.00 32.00 32.00 0.960 ../1dxt/model54.pdb
1 142.22 154.84 127.22 30.00 32.00 34.00 0.958 ../1dxt/model195.pdb
1 171.19 146.19 142.90 32.00 32.00 32.00 0.955 ../1dxt/model94.pdb
1 174.37 131.21 129.78 32.00 32.00 32.00 0.953 ../1dxt/model51.pdb
1 167.50 138.04 134.76 32.00 32.00 32.00 0.952 ../1dxt/model97.pdb
1 157.61 141.37 126.43 32.00 32.00 32.00 0.952 ../1dxt/model60.pdb
1 146.24 136.78 112.96 32.00 32.00 32.00 0.952 ../1dxt/model35.pdb
1 148.56 146.92 122.55 32.00 32.00 32.00 0.951 ../1dxt/model19.pdbç
To test our methodology we build a benchmark formed by 28 simulated docking cases, comprising a wide variety of macromolecular shapes. Each test case consists of a macromolecule atomic structure, five simulated density maps generated from it (at experimental resolutions of 10, 15, 20, 25 and 30Å), and different atomic structures which corresponds to subunit components of the target macromolecule. Each docking test consist in localize the atomic structural subunits into the corresponding complete simulated 3D-EM map. For future developments and comparations the docking benchmark has been made available at download section. Here is the complete list:
|
In each directory (bold face in the list) you can find all the maps in situs format (.sit extension) and the subunits to be docked in PDB format. Since typically are multiple copies of the subunits in the macromolecule map, for comparison purposes, each of them are available in separate pdb files (m1, m2, m3..etc). For example, for running one test using our approach, just use:
Since its a 8-mer, you can find 8 best fits which poses correspond to the original ones (pdb files 1aw5m(1-8) ).