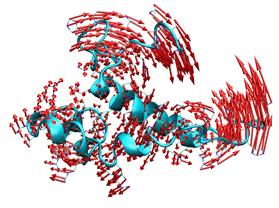
| We actively address the study and simulation of the dynamics of biomolecular systems using Normal-Mode Analysis (NMA), geometric algebra, and other multiscale approximations. The structural flexibility of biomolecules is closely tied to their function, as evidenced by the many conformational changes observed in key cellular processes. Based on the principle "if you know how it moves, you can infer how it works," inferring the intrinsic molecular flexibility from a single conformation can offer direct insights into functional motions. To tackle this problem, we explore novel multiresolution approaches to simulate the dynamics of proteins, nucleic acids, and their complexes. If interested, you can use iMOD, our versatile toolkit for performing Normal Mode Analysis (NMA) in internal coordinates or directly access our iMODS online server. To efficiently explore local protein loop conformations, we incorporate constraints into our NMA formulation to maintain the position and orientation of the loop ends. This novel method, called ilMODE, offers several advantages compared to other methods for concerted loop movements: It has no size limitations. It is highly efficient, as it only requires the diagonalization of a small matrix of size 2N-5, where N is the number of residues in the loop. The elastic network includes both the loop and neighboring protein residues. |
 |