iMOD tips
How are defined Coarse Grained (CG) levels?
|
The Coarse Graining model representation determines both the system mass distribution and the springs network. The available Coarse-Graining models for proteins are listed below:
Next figures illustrate the appearance of the Cα and 3BB2R CG models:
On the left model only Cα atoms (yellow) are considered. On the right, the 3BB2R model (right) have five atoms per residue: three representing the backbone (N, Cα and carbonylic C, in blue, yellow and cyan, respectively), and two representing the side chains (Cβ and a pseudo-atom placed on the center of mass of the remaining side chain, in cyan and red, respectively).
At this moment, only the Full-atom representation is available for nucleic acids:
|
How to use CG models?
To illustrate the usage of different CG models lets try the following examples:
- Computing modes at different CG levels.
- Monte-Carlo at different CG levels.
--------------- Computing desired Normal Modes in different CG models. ---------------
To choose the desired CG model -m option must be added to the basic command followed by model identifier. The basename option (-o) will be used to avoid overwriting of previous re.
3BB2R-> imode 1ab3.pdb -m 1 -o imode3BB2R
You can check the CG models using Jmol: ![]() Ca model and
Ca model and ![]() 3BB2R model.
3BB2R model.
In Ca case, an extra file is produced: imodeCA_ncac.pdb. This PDB file is just a standard PDB containing the coordinates of backbone nitrogen (N), and alpha (Ca) and carbonylic carbons (C), i.e. those atoms needed to define the f and ? dihedral angles. This is the minimum required structure to use our Ca model. A structure with only Ca atoms will not work. In this case, an external software to generate backbone N and C atoms is mandatory. For example, you can use the on-line server: SABBAC.
To animate this modes, the CG model introduced to iMOVE should be the same used in iMODE:
3BB2R-> imove 1ab3.pdb imode3BB2R_ic.evec imove3BB2R_1.pdb 1 -m 1
In Ca case there are less springs than in full-atom. Thus, the amplitude was lowered (-a 0.4). In 3BB2R case it is not neccessary such modification since the number of springs is similar to full-atom.
Its convenient to convert CG models to a full atom representation to avoid problems of compatibility with visualization software:
imove 1ab3.pdb imodeCA_ic.evec imoveCA_FULL_1.pdb 1 -m 0 -a 0.4 --model_out 2
3BB2R->
imove 1ab3.pdb imode3BB2R_ic.evec imove3BB2R_FULL_1.pdb 1 -m 1 --model_out 2
--------------- Monte-Carlo in different CG models. ---------------
To compute MC trajectories:
3BB2R-> imc 1ab3.pdb imode3BB2R_ic.evec -m 1 -o imc3BB2R
As before, to maintain the motion appearance the amplitude was lowered (-a 0.1) in Ca case.
To obtain full-atom trajectories from Ca or 3BB2R modes:
imode 1ab3.pdb -m 0 -o imodeCA --model_out 2
imc 1ab3.pdb imodeCA_icf.evec -m 2 -a 0.1 -o imcCA_FULL
3BB2R->
imode 1ab3.pdb -m 1 -o imode3BB2R --model_out 2
imc 1ab3.pdb imode3BB2R_icf.evec -m 2 -o imc3BB2R_FULL
An extra output file named <basename>_icf.evec will contain the output modes of the corresponding CG level.
How to customize the potential energy model?
For a given structure and CG model, normal modes are determined by its potential energy, i.e. the spring network. In iMODE the potential energy can be customized several ways:
| Functions | Options* | Description |
| Inverse exponential (default) K = k/(1+(x/x0)^p), if x < c, else K=0 |
-P 0 --k0_c 10 --k0_k 1 --k0_x0 3.8 --k0_p 6 |
Distance cutoff (Å) Scale factor Inverse exponential center Power term |
| Simple distance cutoff K = k, if x < c, else K=0 |
-P 1 --k1_c 10 --k1_k 1 |
Distance cutoff (Å) Scale factor |
*Bold options are mandatory.
For example, to use a Ca model with potential energy based on distance dependant functions, type:
imode 1ab3.pdb -m 0 -P 1 -o imodeCA_C
The appearance of this potential energy models with default parameters is shown below. Springs are represented by red segments with thickness proportional to the force constant.
| Inverse exponential (Stiffness inversely proportional to distance) |
Simple distance cutoff (Constant stiffness) |
 |
 |
To customize the potential energy funcitons modify the default options; for example:
imode 1ab3.pdb -m 0 -P 1 --k1_c 5 --k1_k 2 -o imodeCA_C2
Topology and Secondary Structure
iMODE permits the specification of customized inverse exponential functions according to both topology and SS.
The basic command for topology and SS would be:
A functions file (funcTSS.txt) is mandatory. It should conform the following format:
(Note, "#"-begining lines will be omitted)
# SS n k x0 p HH 0 2 3.8 6 HH 1 5 3.8 6 HH 2 3 3.8 6 HH -1 2 3.8 6 EE 0 2 3.8 6 EE 1 5 3.8 6 EE 2 3 3.8 6 EE -1 5 3.8 6 XX -1 1 3.8 6
Each line represents an inverse exponential function. First column specifies the SS. For example, "HH" indicates that both atoms should belong to residues with a-helix SS. By default SS identifiers are: "H" helix, "E" strand, and "C" coil; and any pair of them is allowed. Second column specifies the topology. Here topology represents the sequential distance between two residues; for example, if our protein sequence were ...AGKTLV..., the topology between underlined residues would be 3. Remaining columns define the inverse exponential functional parameters: k, x0 and p, (see the table above).
The wildcards for SS and topology are "XX" and "-1", respectively.
By default, the SS is computed internally, but any user defined SS can be provided using the --ss option:
For example, you can use DSSP to compute SS (1ab3.dssp) and with the aid of this simple perl script you can convert it into our SS file format (1ab3.ss)
Our SS file format (1ab3.ss) is a simple two column ASCII file. The first column corresponds to the sequence index, and the second one to a single character SS identifiers.
Customize potential energy by topology only.
The basic command for topology is:
The topology functions file (funcTOP.txt) is:
# SS n k x0 p XX 0 2 3.8 6 XX 1 9 3.8 6 XX 2 5 3.8 6 XX 3 3 3.8 6 XX 4 2 3.8 6 XX -1 1 3.8 6
Customize potential energy by SS only.
The command to take into account SS only would look like this way:
The functions file (funcHE.txt) is:
# SS (j-i) k x0 p HH -1 2 3.8 6 EE -1 5 3.8 6 HE -1 3 3.8 6 XX -1 1 3.8 6
This file applies different functions to atom pairs belonging to residues with SS: H vs. H (HH), E vs. E (EE) and H vs. E (HE). The XX function will be applied to remaining pairs of atoms.
Given the non-Helical or non-Sheet regions are more flexible than the rest, some times may be interesting to increase flexibility in those regions. Corresponding command would be:
The functions file (funcCX.txt) is:
# SS (j-i) k x0 p CC -1 0.2 3.8 6 XX -1 1 3.8 6
The springs associated to last example are represented below. First and second function springs are colored in orange and red, respectively. For clarity bo th kinds of springs were shown with similar thickness.
 |
--------------- User custom potential. ---------------
The user can define its own potential thought a file using the -K option. Type at the command prompt:
This ASCII file (imodeKi_Kfile.dat) has three columns to define the force constants (K) for each atomic pair:
1 2 9.962940E-01 1 3 9.292819E-01 1 4 6.086772E-01 1 5 9.329874E-01 1 6 8.701856E-01 ...........
Each line represents one spring. The first and second colums are the atomic indices (begining with 1), and the third is the force constant.
To obtain a Kfile template for your macromolecule that you can adapt to your convenience, use:
The resulting file, imodeKc_Kfile.dat, will contain the force constants for current potential energy model, in this case, the simple cutoff model.
How to deal with huge systems?
- Fixing custom ICs.
- Fixing by secondary structure
- Fixing ICs randomly.
The capsid of the Cowpea Chlorotic Mottle Virus (CCMV) is a huge protein structure composed of 180 chains, 28620 aminoacids and 214440 atoms. In terms of ICs it means about 58000 ICs: 56994 dihedral angles (f and ?) and 1074 inter-chain rotational/translational variables. Perform NMA with 58000 ICs it's impractical in standard PC box, since there would be needed a 64-bit computer with about 30Gb of RAM memory, and each diagonalization step would last even days. To overcome this we are going to fix the 90% of dihedral angles while keeping mobile all rotational/traslational degrees of freedom:
imode> imode> Welcome to the NMA in Internal Coordinates tool v1.10 imode> imode> Reading PDB file: 1cwp_prot.pdb molinf> Protein 1 chain 1 A segment 1 residues: 149 atoms: 1122 molinf> 2 B segment 1 residues: 164 atoms: 1226 molinf> 3 C segment 1 residues: 164 atoms: 1226 molinf> Protein 2 chain 1 A segment 1 residues: 149 atoms: 1122 ................................................................... molinf> Protein 60 chain 1 A segment 1 residues: 149 atoms: 1122 molinf> 2 B segment 1 residues: 164 atoms: 1226 molinf> 3 C segment 1 residues: 164 atoms: 1226 molinf> SUMMARY: molinf> Number of Molecules ... 60 molinf> Number of Chain ....... 180 molinf> Number of Segments .... 180 molinf> Number of Groups ...... 28620 molinf> Number of Atoms ....... 214440 molinf> imode> Coarse-Graining model: Full-Atom (no coarse-graining) imode> Selected model residues: 28620 imode> Selected model (pseudo)atoms: 214440 imode> Number of Dihedral angles: 55920 imode> Number of Inter-segment coords: 1074 (Rot+Trans) imode> Number of Internal Coordinates: 56994 (Hessian rank) imode> Input CG-model Fixed Internal Coordinates: 50328 imode> Input CG-model Final Internal Coordinates (sizei) = 6666 imode> Range of computed modes: 1-20 imode> Creating pairwise interaction potential: imode> Inverse Exponential (16245240 nipas) cutoff= 10.0, k= 1.000000, x0= 3.8 imode> Packed-Hessian/Kinetic matrices mem.= 177.769 Mb (rank= 6666) imode> Fast Hessian Matrix Building O(n^2) [hessianMFAx()] 17.24 sec imode> Fast Kinetic-Energy matrix Building O(n^2) [kineticMFAx()] 20.34 sec imode> Eigenvector matrix mem. = 1.067 Mb imode> Diagonalization with XSPGVX()... 380.06 sec imode> Showing the first 10 eigenvalues: imode> imode> MODE EIGENVALUE imode> 1 1.72062e-04 imode> 2 1.75517e-04 imode> 3 1.77022e-04 imode> 4 1.79158e-04 imode> 5 1.81447e-04 imode> 6 3.30420e-04 imode> 7 3.39147e-04 imode> 8 3.44462e-04 imode> 9 3.57581e-04 imode> 10 3.65270e-04 imode> imode> imode> SAVED FILES: imode> Log file: 1cwpDH09.log imode> Model PDB: 1cwpDH09_model.pdb imode> Fixation mask file: 1cwpDH09.fix imode> ICS eigenvectors: 1cwpDH09_ic.evec imode> imode> Bye!This way, only 6666 ICs were considered. Below you can check 5th and 18th normal modes, from the H and A and irreducible representations, respectively. On the left, the flash movies, on the left the displacement vectors.
 |
|
 |
 |
 |
imorph ccmvmorph_fitted.pdb ccmv_swln_1_full.vdb -i 300000 -r 1 -n 0.99999 --morepdbs --prob plain -o ccmvmorph2 In first step we added: -e 0.05 to speed up convergence, --nowrmsd to disable the Weighted-RMSD alingment (which is not suited to huge motions), and --more_pdbs to force saving the fitted structure. Note this fitted structure was still about 5 Å Ca RMSD away form target. In a second refinement step, we used all available modes (-n 0.99999) with a different mode selection scheme (--prob plain). Since we were closer to the target structure, the --nowrmsd option was removed. Note that to minimize NMA time in both steps only the rotational/translational ICs were considered (-r 1). Now a successful morphing result is obtained. It's only 0.95 Å Ca RMSD from target structure. You can check its beautiful movie below.
How to fix ICs?
- Randomly (commented before Q4)
- Un-fix χ dihedral angle.
- By Secondary Structure.
- Customized IC subset.
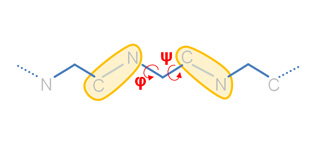
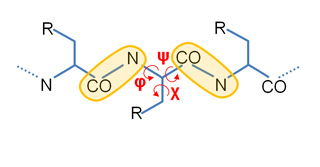
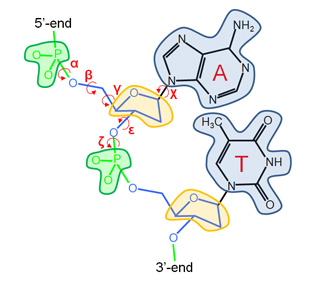
Our Internal Coordinates are: φ, ψ and χ dihedral angles for proteins, and α, β, γ, ε, ζ and χ for nucleic acids (see figure below). Another six additional rotational/traslational degrees of freedom are added every extra chain
| Protein | RNA |
 |
 |
By default we consider all but χ. The removal of this angles does not affect the low energy modes and will reduce the problem dimensionality around 1/2 in proteins and 1/5 in nucleic acids.
With iMOd tou can fix the ICs depending on the secondary structure SS it belongs to. This can be done using the −−ss option followed by a letter identifiyer (H for helix, E for beta strands and C for others). Note you may fix any combination of SS by adding an identifier as you need; for example, "−S EC" will fix beta and coil. To fix all ICs related to α-helices and β-sheets, just type prompt:
By default, the SS is computed internally, but any user can defined it own SS throught a file and using the −−ss option:
For example, you can use DSSP to compute SS (1ab3.dssp) and with the aid of this simple perl script you can convert it into our SS file format (1ab3.ss)
Our SS file format (1ab3.ss) is a simple two column ASCII file. The first column corresponds to the sequence index, and the second one to a single character SS identifiers.
Finally, you can fix any domain/s or region/s of your macromolecular structure. Any IC set can be considered fixed using the −f option with a mask file as;
where the mask file imodeFIX.fix is an ASCII file with four columns. The first column is the residue index (begining with 0), and the rest represents the φ, ψ and χ dihedral angles, respectively. In nucleic acid case, the file will have seven columns instead of four, to account for the (six) α, β, γ, ε, ζ and χ dihedral angles.
A dummy mask (fully mobile) can be obtained using the −−save_fixfile option in iMODE program.
such mask looks like:
0 0 0 1
1 1 0 1
.........
276 1 0 1
277 0 0 1
278 1 0 1
.........
522 1 0 1
523 0 0 1
To customize just change "1" by "0" for fixing ICs, save it, and use with imode with −f option to load it and restric the NMA only with the non-fixed ICs as variables.
The extra zeros at some lines account for residues lacking corresponding ICs, i.e. Glycines and Alanines (no χ), and Prolines (no φ and χ). Note that the −x option must be added on previous commands if you plan to keep mobile some χ dihedral angles. If the macromolecule had several chains, six inter-chain rotational/traslational ICs are added: three x, y and z traslations and three rotations around x, y and z axis, respectively. For example, if there is a new chain after residue 187 (index 186) the mask file will be:
.........
186 1 0 1
187 1
187 1
187 1
187 1
187 1
187 1
187 1 0 1
.........
Be free to include protein, DNA, RNA and rigid ligands altogether
GroEL is composed of three domains: apical (top red), intermediate (cyan), and equatorial (bottom red). The apical domain interacts with folding intermediates and GroES, the equatorial domain hydrolyzes ATP, and the intermediate domain is flexible and connects these apical and equatorial domains.
 |
To obtain a successful fitting the structure should be maintained as flexibile as possible, i.e. if we fix very much the structure will be very rigid and the target conformation will not be addressed. To this end the apical and equatorial domains will be fixed while keeping the intermediate domain fully flexible. In the mask file imodfitDOM.fix only the ICs belonging to regions of the intermediate domain are keept mobile: 136-193 and 331-409 (indexed from 1 to 524). Further information about the mask file generation is provided in Q1 within this FAQ.
The output is the following:
imodfit> imodfit> Welcome to iMODFIT v1.28 imodfit> imodfit> Model PDB file: 1aon.pdb molinf> Protein 1 chain 1 segment 1 residues: 524 atoms: 3847 molinf> SUMMARY: molinf> Number of Molecules ... 1 molinf> Number of Chain ....... 1 molinf> Number of Segments .... 1 molinf> Number of Groups ...... 524 molinf> Number of Atoms ....... 3847 molinf> imodfit> Coarse-Graining model: Full-Atom (no coarse-graining) imodfit> Selected model number of residues: 524 imodfit> Selected model number of (pseudo)atoms: 3847 imodfit> Target Map file: 1oel.ccp4 imodfit> Best filtration method: 2 FT(x10)=0.090s Kernel(x10)=0.080s imodfit> Number of Inter-segment coords: 0 (Rot+Trans) imodfit> Number of Internal Coordinates: 1033 (Hessian rank) imodfit> Input CG-model Fixed Internal Coordinates: 760 imodfit> Input CG-model Mobile Internal Coordinates (size) = 273 imodfit> Range of used modes: 1-54 (19.8%) imodfit> Number of excited/selected modes: 1(nex) imodfit> imodfit> Iter score Corr. NMA NMA_time imodfit> 0 0.336409 0.663591 0 0.73 sec imodfit> 199 0.324457 0.675543 1 0.50 sec imodfit> 379 0.285427 0.714573 2 0.49 sec ................................................. imodfit> 12857 0.080700 0.919300 14 0.50 sec imodfit> 50000 0.064124 0.935876 END imodfit> imodfit> Movie file: imodfitDOM_movie.pdb imodfit> Final Model: imodfitDOM_fitted.pdb imodfit> Score file: imodfitDOM_score.txt imodfit> Log file: imodfitDOM.log imodfit> imodfit> Success! Time elapsed 00h. 16' 18'' imodfit> Bye!
The solution is only 3.77 Å Cα RMSD from the target structure used to simulate the map. This is a good result for a structure with 75% of its ICs fixed. Note that, to obtain a good result with such a fixed structure, the number of iterations was increased (-i 50000). If you increase the number of iterations even more you will reach lower RMSD.
You can play interactively the trajectory movie in ![]() Jmol, check the fitted structure
Jmol, check the fitted structure ![]() imodfitDOM_fitted.pdb and watch the flash movie below. For illustrative purposes the fixed domains were colored in red.
imodfitDOM_fitted.pdb and watch the flash movie below. For illustrative purposes the fixed domains were colored in red.